With the revised Annex 1 coming into effect last year, the pharmaceutical industry continues to strategize the best way to attain and maintain compliance with the intensive regulation on sterile medicinal products. Aseptic fill-finish manufacturing, for its part, is established on precise science and the highest standards of safety. While much of the science, technology, mechanisms, and regulations behind parenteral drug products are complicated and nuanced, the net goal is simple: to fill a container with drug product in a way that ensures product integrity and patient safety. How the industry and stakeholders collectively work towards this goal is predicated on best practices, technological implementation and innovation, and robust regulatory frameworks. Central among those frameworks is EU GMP Annex 1 with the following stated purpose:
“The manufacture of sterile products covers a wide range of sterile product types (active substance, excipient, primary packaging material and finished dosage form), packed sizes (single unit to multiple units), processes (from highly automated systems to manual processes) and technologies (e.g. biotechnology, classical small molecule manufacturing systems and closed systems). This Annex provides general guidance that should be used in the design and control of facilities, equipment, systems and procedures used for the manufacture of all sterile products applying the principles of Quality Risk Management (QRM), to ensure that microbial, particulate and endotoxin/pyrogen contamination is prevented in the final product.”
As with any new or updated regulation, there were plenty of questions to greet the new Annex 1 upon its adoption. Why did this version expand so significantly upon its predecessor? What’s the full scope of the regulation as it stands, and what are the expectations around compliance? How should US manufacturers view Annex 1? And how should the increased emphasis on robotics, automation, and barrier technologies be viewed?
Annex 1: A Story of Collaboration
Pharmaceutical manufacturing is an internationally oriented and interconnected industry. Technologies, innovations, breakthroughs, and collaborations can come from every corner of the globe. It’s not uncommon for smaller-sized manufacturers, who excel at producing any number of precise technologies, to serve regular international customers. In the larger context of treatment and patient care, one of the great unifiers in our world is the ability of multiple countries, governmental groups, organizations and companies to come together in the fight against illness and disease. The drafting process of the new Annex 1 reflected this same spirit as regulatory bodies, think tanks, and experts from all over the world provided recommendations and feedback for what content should be included in the document. All in all, the document presents the view of the European Commission, the international PIC/S (Pharmaceutical Inspection Convention & Pharmaceutical Inspection Co-operation Scheme), and the World Health Organization, and is the most comprehensive, collaborative regulatory document ever authored. The PIC/S alone includes over 56 regulatory authorities, including the US Food and Drug Administration.
And while it is an EU regulation, it reflects US standards and the criteria inspectors will be evaluating, as evidenced by the FDA’s participation and explained at a recent ISPE panel discussion with regulatory heads of the FDA, EMA and MHRA.
An Expanded Principle
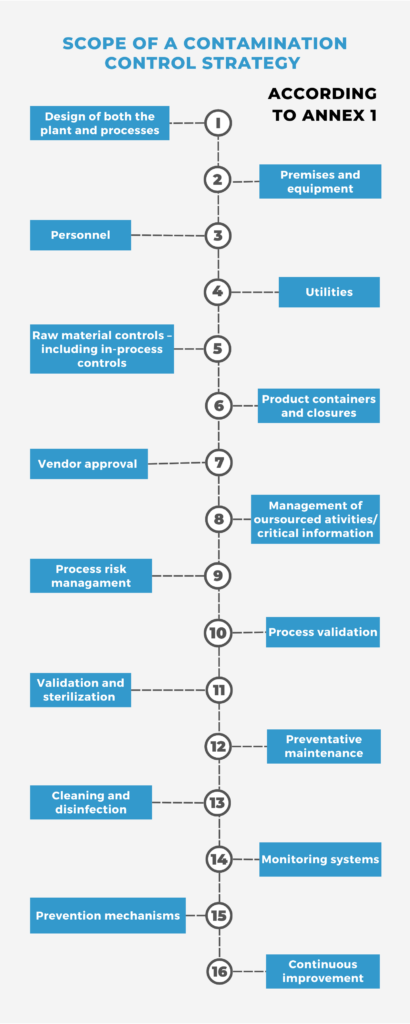
The 2023 Annex 1 was not only significantly expanded in length (58 pages compared to 16 pages from the 2008 version) but also introduced a new structure (categories, chapters, document map, and a glossary of terms) and in-depth coverage of any areas directly related to manufacturing sterile products. The principle, in seven detailed sections, highlights the special requirements needed to minimize contamination and calls for a comprehensive Quality Risk Management approach (when identifying and evaluating any risk to quality), and a Contamination Control Strategy that “…should establish a robust assurance of contamination prevention.”
In greater detail, Annex 1 encourages the use of “appropriate technology”, specifically mentioning isolators, RABs, and robotic systems—a significant choice by the authoring committee and an indication of the superiority of these solutions as it pertains to sterility assurance and quality management.
The in-depth principle is a departure from and improvement on its predecessor and informs the guidance throughout the rest of the document, including premises, equipment, production & specific technologies, and environment/process monitoring.
Critical Themes:
Contamination Control Strategy
Contamination Control Strategy (CCS)is a term introduced in the revised Annex 1 and is defined in the regulation as:
“A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.”
Pharmaceutical manufacturers, of course, had many of the elements of CCS built into their own Quality Management System that were informed by various risk assessment protocols. But in including it as a comprehensive regulatory concept, Annex 1 put forth the most stringent, harmonized approach for product safety and integrity ever by a regulatory presence.
First Air
The revised Annex 1 emphasizes the proper configuration and use of unidirectional airflow and spotlights the importance and priority of the “First Air” principle. These terms are defined in the Annex as:
- First Air – Air that has not been interrupted prior to contacting exposed product and product contact surfaces with the potential to add contamination to the air prior to reaching the critical zone.
- Unidirectional airflow – An airflow moving in a single direction, in a robust and uniform manner, and at sufficient speed, to reproducibly sweep particles away from the critical processing or testing area.
As undisturbed unidirectional airflow is a product’s final defense against contamination, Annex 1 emphasizes these concepts in an operation’s design, contingencies, and application. Tools like CFD analysis and smoke studies should be used to carry out airflow studies and demonstrate the effectiveness of your unidirectional airflow approach. Annex 1 calls for both “at rest” and “in operation” studies in any critical zones where air movement could be problematic and corresponding design improvements where needed. For fill-finish operators, maintaining aseptic technique in whatever configuration chosen is paramount. When using robotics and automation, simplified, proven designs that minimize airflow turbulence are key to meeting the threshold set out by this regulation.

Barrier Technology
Barrier technology—specifically isolators—have seen steady growth in utilization over the past ten years as their efficacy in providing an ideal, low-risk, Grade A environment for drug production continues to be demonstrated. And as the adage goes: “With great power comes great responsibility.” Or, if you prefer the compliance version: With great use comes great regulation. The new Annex 1 clearly reflects the growing application and necessity of isolator technology as it mentions it specifically 48 times (compared to 15 times in the 2008 revision). And though regulatory compliance isn’t always easy (nor should it be), that the largest coalition of regulatory experts and organizations, through Annex 1, encourage greater use of isolator technology is notable. Annex 1’s expanded emphasis on isolators and recommended guidelines on their safe and effective utilization is a clear indication of where the pharmaceutical manufacturing industry is headed.
Environmental Monitoring
Environmental monitoring is expanded on in the new Annex 1, including a detailed outline of what should be included in an Environmental Monitoring program (non-viable particle, viable particle, temperature, RH and aseptic process simulation). In addition to the environmental monitoring program, Annex 1 now recommends continuous monitoring as another means of risk reduction. For total particulate monitoring (non-viable), it calls for an active flow sampler to capture “all interventions, transient events and any system deterioration.” And for Viable, continuous monitoring should be carried out in Grade A areas and should be operational during “for the duration of critical processing, including equipment setup…”
Automated Solutions
Much like isolator technology, automation saw increased attention in the new Annex 1. Automation was explicitly highlighted for its ability to de-risk certain interventions and is encouraged anywhere it can reduce the likelihood of decontamination. It’s also highlighted for its potential role in visual inspections and rapid technology possibilities for environmental monitoring. When combined with another key concept in Annex 1, designed engineering solutions, the regulation actively encourages manufacturers to explore the advantages of automation, both within a CCS and the larger context of a quality management.
Prioritizing Manufacturing Efficiency and Regulatory Compliance

AST’s aseptic processing systems are not only built with an eye towards efficiency and innovation; they’re designed to be intuitive tools that are regulatorily robust and forward-looking. As pioneers in the use of robotics in the aseptic fill-finish space, we provide customers with the technological solutions, service, and expertise needed to navigate all aspects of the drug development process, from project planning to aftermarket care and every manufacturing and regulatory uncertainty in between.
Annex 1 represents tremendous progress towards harmonizing regulations across the pharmaceutical manufacturing industry and codifies the crucial roles that automated and barrier technology play in the safety and quality of medicinal production.